The AI for materials space has been picking up a lot of steam—materializing, even. With frontier labs Periodic, Cusp, Radical, Google DeepMind, and Lila focusing heavily on materials, national labs kicked into high gear with Project Genesis, and broader momentum in AI for science, the dream of autonomous materials discovery seems to be on the near horizon. And it truly would be a dream. The rapid discovery and improvement of materials could lead to revolutions across compute, energy, advanced materials and usher in a new era of human abundance.
However, many breakthroughs in research have had major caveats, materials discovery has a wasteland of failed early stage companies, and many of the frontier lab startups have struggled to produce tangible results and are shifting focus to R&D SaaS or narrowing their materials problem space.
My read after spending time working on ML for materials is that the researchers in the space are treating materials discovery as a search problem, working on generative models that can propose new materials with desired properties and self-driving labs that can rapidly carry out experiments. These approaches will not be enough. Although high-throughput and data-driven methods may have a part to play, when I think of breakthroughs in Li-ion batteries, 2D materials, perovskites, and catalysis, they took years of meticulous, collaborative, creative research where insight was slowly built up through the scientific method.
Experimental throughput was never the bottleneck to scientific discovery—if it was, why didn’t every national lab and materials/chemistry R&D company like Shell, Corning, 3M, Applied Materials just hire dozens of cheap technicians per team, 100x the experiments they’re able to run, have a PI and data scientist look at trends in the results and instruct on follow-up experiments?

Maybe it’s the risk profile of these large labs, but a more likely explanation is it’s not the throughput of experiments being the bottleneck to materials innovations. It’s that it takes intelligence to do rigorous scientific research, to build new theories and models of the world based on experiments that help guide further experiments and discover new materials, and that intelligence is the bottleneck.
However, the key trend here, as is the key trend behind almost everything the past few years, is the rise of LLMs. Intelligence is shifting from a bottleneck to a commodity, and this is why the autonomous materials discovery space is suddenly so interesting. A high-throughput robot lab following an optimization algorithm based on a performance metric has way less potential than a high-throughput robot lab controlled by an AI materials scientist carrying out the scientific method. This is indeed the vision of the most forward-thinking neolabs, but there is still much work to be done.
Below is a first-principles breakdown of autonomous materials discovery and what I believe to be a particularly interesting and high-leverage open problem—autonomous characterization (will define soon, for the non-materials scientists).
The Scientific Method for Materials
Materials innovations have brought us everything from semiconductors to lithium-ion batteries to solar cells, and even vulcanized rubber—discovered by Charles Goodyear in 1839 when, after years of searching for a way to make rubber durable, he accidentally left a mixture of natural rubber and sulfur on a hot stove.
Although the term “materials discovery” evokes a picture like that of Charles Goodyear combining things in a new way and suddenly creating a vastly superior material, the materials research process that brought about all our modern technologies is much more meticulous and rational.

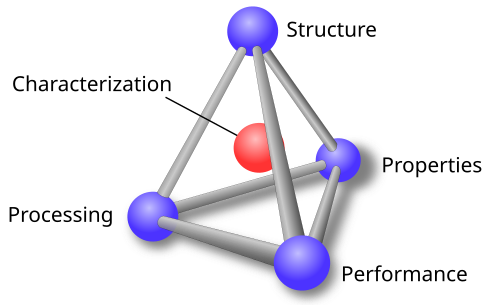
Modern materials science can be broadly divided into theory/computation, synthesis, and characterization.
- Theory and computation uses physical models and physics-informed simulations to explain why materials behave the way they do and to suggest promising new compositions and processing techniques.
- Synthesis is the process of actually making the material, whether by chemical growth, aqueous precipitation, thin-film deposition, or other fabrication methods.
- Characterization measures what was made, using x-rays and microscopes to probe a material’s structure, composition, and properties to determine whether the synthesis succeeded and whether the material behaves as theory predicted.
The feedback loop between these 3 components is what forms the scientific method for materials science. The existing understanding of the world modeled by theory/computation (“priors”, if you will) is used to create hypotheses about what might work, the experiment to synthesize the material is then run, and the sample is characterized to provide a holistic and mechanistic view of the results, which is then used to update our models and formulate new hypotheses.
Walking through some concrete examples of these components in action might be helpful. A recent paper from the Chueh group at Stanford discovered a way to electrochemically induce disorder in battery cathodes to prevent lattice collapse, preserving microstructural integrity and extending cycle life.
Previous experiments / literature (where structural insights were derived from characterization) had shown that a major of performance degradation in lithium-ion batteries was that the layered cathode structure shrinks/collapses when deeply delithiated.
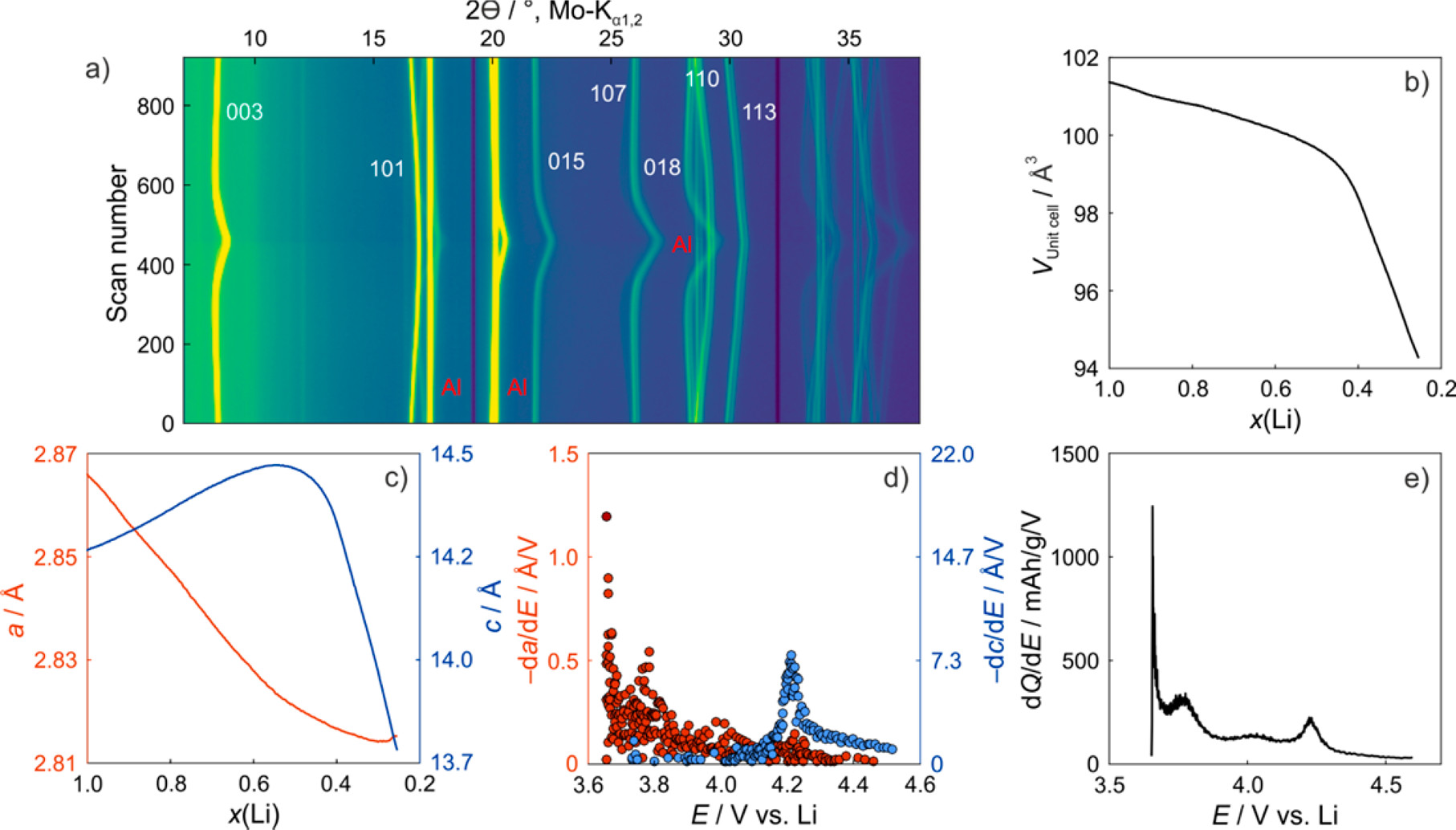
The Chueh group hypothesized that introducing partial disorder between the transition metals and lithium in the lattice would reduce the collapse in the cathode. They synthesized samples where they attempted to introduce this partial disorder through a redox process. These samples were then characterized using operando XRD and XAS, revealing significantly less lattice collapse during cycling and metal oxidation state changes indicating the release of O2 and structural disordering, with trends matched to the cycle life and synthesis parameters. The result of careful analysis confirms a new technique to improve battery stability and adds to the body of theory.
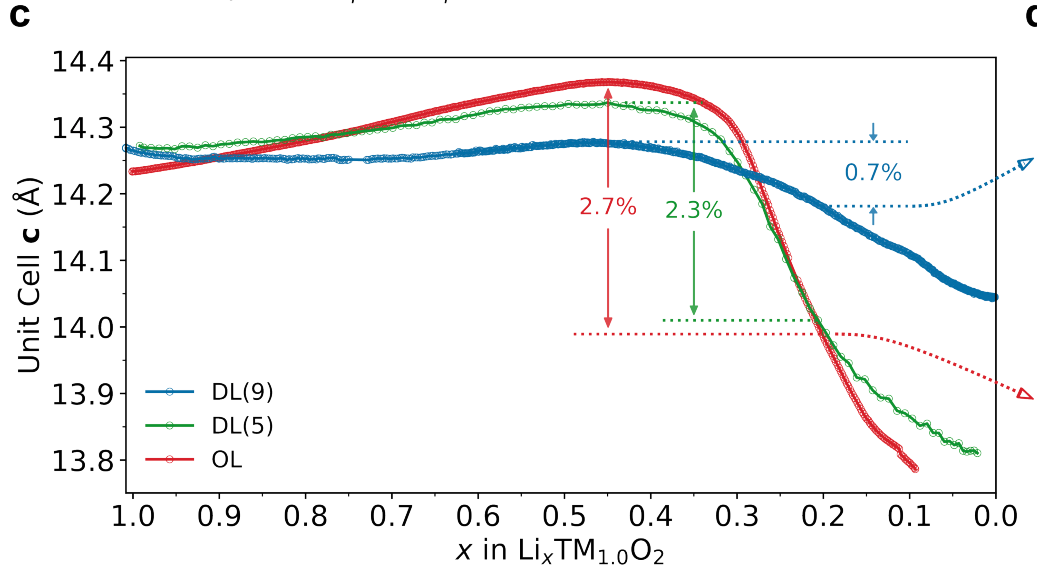
Characterization data from that confirmed electrochemically induced disorder.
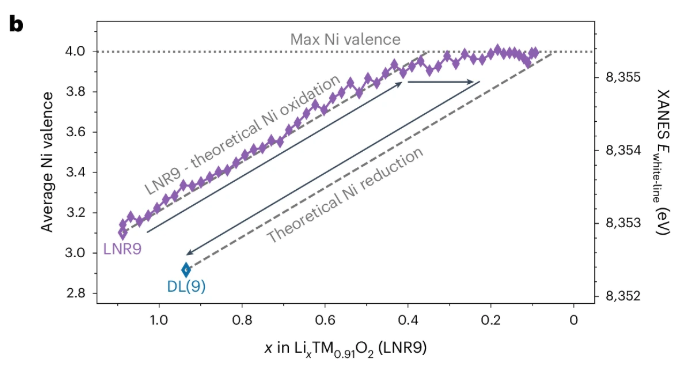
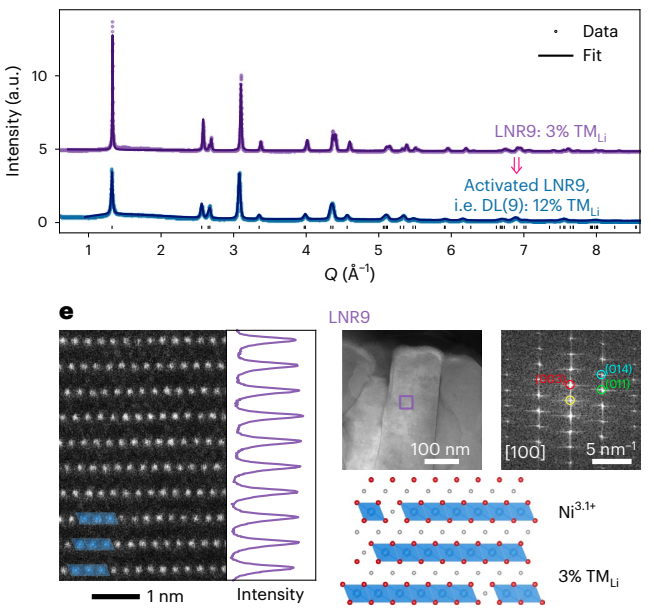
Hundreds of examples of discoveries driven by a similar process can be found in the development of any advanced material. For example, MoSe2 is a 2D transition metal dichalcogenide material with applications in transistors, batteries, CIGSe solar cells, catalysis (hydrogen evolution), and much more. Looking through a review of its developments and applications, one sees a plethora of referenced papers developing new insights, applications, and synthesis/processing techniques— the first synthesis of vertically aligned layers, the observation of direct bandgap emerging in monolayers, investigation of properties in cleaved CIGSe solar cells (something I helped with), etc. Each of these steps forward required cycles of the scientific method.
In order to build AI that can do science, it must be able to carry out the scientific method, which in the case of materials means theory, synthesis, and characterization to the level demonstrated in the above examples. Characterization in particular is key to unlocking crucial information about the relationship between structure, properties, processing, and performance.
Current Focuses in Autonomous Materials Discovery, and Why It Won’t Work
My perspective is that the ML for materials community seems too focused on the theory/computation and synthesis parts of the loop, treating materials discovery as a search problem over the materials space and synthesis parameters rather than a research problem.
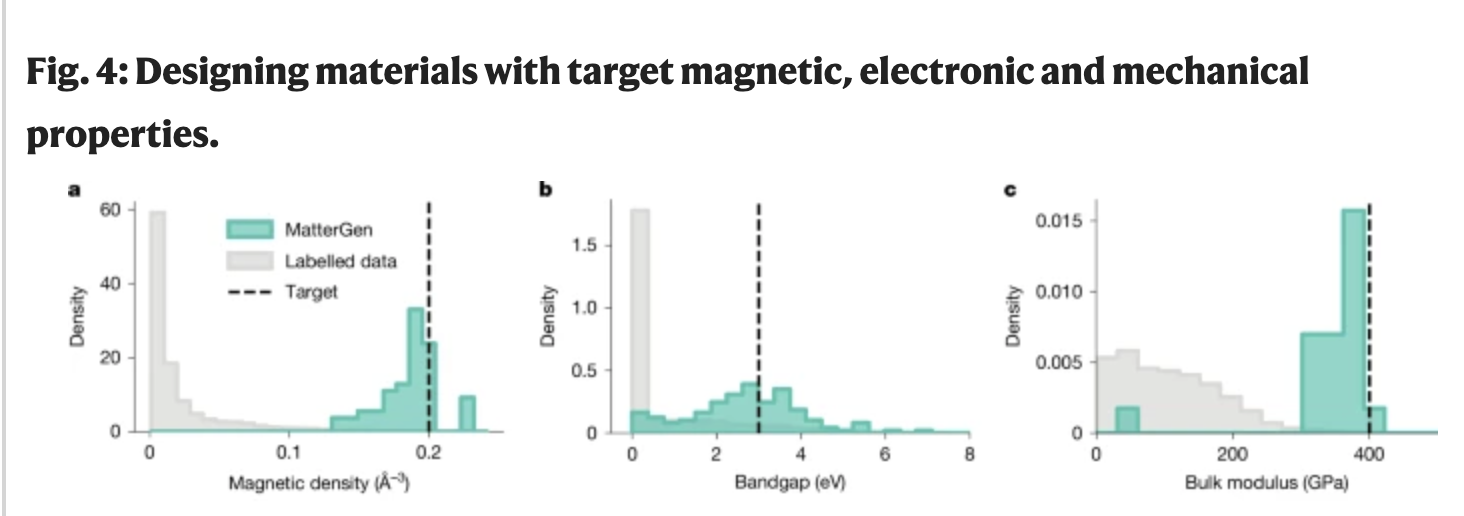
There’s a lot of attention in the literature on inverse design, proposing potential candidate materials with desired properties using generative models. This is seen as a leap forward from traditional methods like screening, but the ability of conditional generation to generate materials with desired properties is still far from robust (see the spread in property values below from a leading materials diffusion model). We’re quite far from the ideal of a model instantly proposing a new perfect material with ideal properties. More importantly, the process of actually synthesizing and scaling up the desired material is another layer of difficulty on top of that. This is because bringing materials from models to the real world requires real-world experimentation and iteration.
There’s also a lot of attention on self-driving labs, key to unlocking high-throughput; they should be able to run experiments faster, for longer, and with greater precision than human scientists. The current rhetoric touts the ability to rapidly explore large chemical search spaces, finding optimal synthesis ingredients and parameters to discover materials with desired properties. However, the “intelligence” driving this search is usually bayesian optimization or some basic data-driven active/reinforcement learning approaches. This approach optimizes for a performance metric, taking in scalar values of results/parameters from past experiments and deciding what the synthesis parameters of the next experiment should be. It’s very data-driven, but not very intelligent—with only surface-level measurements of each sample, you don’t build an understanding of what’s really going on in a sample and why it performs the way it does. There have been some attempts that integrated some basic characterization in the loop, but they are limited in a variety of ways and still don’t escape the mindset of building search algorithms rather than autonomous researchers. (There’s more to be said, see footnote [2])
Trying to create a better material with these optimization techniques without extensive characterization and analysis is like trying to escape a maze by running fast instead of reading a map. Given the above examples of how much characterization, analysis, and rationality went into the discovery of new materials, it should be clear that these optimization algorithms and proposal generators are far from achieving real materials innovations.
Perhaps the frontier AI for materials startups are best equipped to really bring intelligence into the loop and build AI and autonomous labs that can actually do science. They understand the skyrocketing potential of LLMs, and that given the right context and post-training, multimodal LLMs will definitely be able to make many of the decisions that materials scientists make. Accordingly, their goals are to create “AI scientists” and “scientific method machines” as well as the autonomous labs to allow the AI systems to run real-world experiments.
However, I don’t believe LLMs are all you need to build AI scientists. There’s this idea in frontier AI that verifiable rewards means LLMs can be post-trained in RL environments to complete any task. The asymmetry of verification and verifier’s rule is why coding, competition math, benchmarks, etc are so effectively conquered by AI. And to some degree, materials science is verifiable—a material’s properties can be measured and its merit scored. This is an argument for the viability of AI materials scientists often made to the more VCs and the more AI-pilled silicon valley crowd, but there are caveats to materials science’s verifiability and scalability that don’t apply to things like coding (and even bio, see footnote [3]). Simulations are inaccurate, the data layer is much more difficult, and real-world experiments are noisy with much sparser reward signals due to the time and resources it takes to run physical experiments. You can’t treat the lengthy process of iteratively and adaptively going through the scientific process to create a new discovery as 1 verifiable task.
Even with LLMs, I believe you need to break down materials discovery into the pieces of the scientific method and train bespoke tools and models. Lila Sciences seem to share this view. They’re building a central LLM agent with access to custom MCPs, including tools like MLIPs, literature search, and characterization interpretation tools. But however much attention is being put into training models for characterization interpretation, it probably isn’t enough. The upside to unlocking the characterization step of the loop is so high, and we are quite far from reaching its potential.
The Characterization Bottleneck
They say eyes are the window to the soul. In that vein, X-rays and electrons might be the window into a material’s soul.
Characterization is so incredibly important to materials research. Billion-dollar synchrotron light-sources are prized research facilities around the world. Lab-scale instruments like Raman, XPS, SEM are found in almost every research university. Thanks to these tools, we’re able to decipher the locations of atoms, see grain boundaries and defects, understand the arrangement and energies of electrons, and so much more. These are things we’d never be able to know just by looking at a piece of silicon or slab of gold.
As a quick briefing, characterization techniques involve probing material samples with beams of light (photons), electrons, or other particles to either generate magnified images to see the sample directly or spectra with peaks encoding information composition, structure, and physical properties. The imaging techniques fall under microscopy, including techniques like optical microscopy, electron microscopy (SEM, TEM, EELS), and scanning probe microscopy (AFM, STM). The measurements resulting in spectra fall under spectroscopy and scattering/diffraction, resulting from techniques like XPS, XAS, XRD, Raman, NMR, etc (where the X stands for X-rays). Characterization isn’t just taking these measurements, but also the process of building a mechanistic picture of a material from multiple imperfect, complementary signals, and that interpretation arguably requires the most expert “tacit knowledge” in the materials discovery loop. Analysis of characterization data often takes weeks or months, and many prominent materials scientists focus their entire careers on this analysis.

AI for characterization needs to take in characterization spectra and/or images as well as experimental/literature context to generate reasonable proposals for the material structure. Then from the structure, relationships to performance and properties can be understood. Interpreting microscopy images seems fairly straightforward with how good general vision-language models have become. Spectra, on the other hand, will likely require bespoke models and architectures beyond LLMs, the same way LLMs would never be able to replace MLIPs (machine-learned interatomic potentials, one of the big successes of computational materials science, able to quickly calculate the energy and forces on an arrangement of atoms), which require GNNs and swaths of DFT-calculated atomistic energy data.
Building architectures to make major improvements to spectral inversion is not an easy problem, and I’ve heard of many bright materials researchers spending years on this before deciding it’s too difficult and shifting focus.
The Path to Autonomous Characterization
Though the task is difficult, there are believable paths. I’ve been exploring and testing promising approaches to tackle the most interesting and high-leverage use cases. Here I’ll explain the landscape of existing work and problems I am working on.
Traditional ML property inference— Most research over the past few years did something along the lines of featurizing spectra and predicting properties like coordination number, mean bond distance, chemical species, etc depending on the type of spectroscopy. These approaches are generalizable to different types of spectroscopy and predicted properties, but are fairly rudimentary and only give a partial picture.
ML-Assisted Pattern Matching— There have also been some approaches that pattern match the spectra of known/proposed structures with the target spectrum, potentially aided by ML. Pattern-matching/fingerprinting have been staples of traditional spectroscopy analysis, but even with ML, these approaches are limited by search space and may suffer from confirmation bias (in the structure set and/or training data).
Conditional Diffusion Models — These approaches get closer to the idea of generating structures that satisfy the condition posed by a target spectrum. Materials diffusion models like Mattergen, DiffCSP, and CDVAE have been able to generate materials conditioned on target properties, so it’s a natural direction to try extending these to spectra. There has been some limited success, notably with XRD, but these models are far from robust and scalable. The use of XRD is also largely limited to crystalline structures and can’t resolve more complex systems with defects, vacancies, or amorphism.
I spent some time trying to use conditional diffusion approaches for XAS (X-ray Absorption Spectroscopy) spectrum -> structure generation. XAS is one of the most useful techniques for understanding complex, functional materials like batteries and catalysts, but quite difficult to interpret quantitatively. Unlike XRD, calculation of a spectrum given a structure requires on the order of hours for a material, so verification is more difficult. Guided diffusion techniques thus also require an ML surrogate for the forward calculation that is accurate even on unrealistic/noisy structures, which is non-trivial since XAS training data is usually stable structures. This paper showed a proof of concept using guided diffusion on a very simple carbon system, but no one has demonstrated a more robust and general XAS spectrum to structure inference. I spent a few months trying and struggling with classifier-free guidance and a variety of more involved diffusion guidance techniques like DPS, UG, and AdamDPS using a Mattergen backbone. I’m now working on workflows to train more robust forward models for structure -> XAS inference, which will hopefully enable guided diffusion or MLIP-based methods for XAS interpretation.
MLIPs + Optimization Algorithm— There have also been interesting MLIP-based approaches that use MLIPs in conjunction with an optimization algorithm like gradient-descent or Monte Carlo methods to find structures that both satisfy the spectrum constraint and are also energetically stable. This is not unlike the guided diffusion approaches, except instead of a diffusion prior and its more finicky time-step specific gradients guiding towards a more stable material, an MLIP “should” work with any arrangement.
This approach seems promising, especially after my struggles with the strict spectrum guidance pushing the generation off-manifold leaving the diffusion prior unable to generate a stable material. However, gradient descent and diffusion guidance are limited by the smoothness of the loss landscape. Any optimization approach (and perhaps the diffusion approaches as well) are also limited by the ceiling of quantitative interpretation of these spectra. Calculated XRD and XPS spectra, for example, can be calculated easily and align well with experiment, while calculated XAS (through approaches like FEFF) often don’t match up perfectly with experiment. Thus, approaches that use an ML forward model for XAS to assess/guide generations may not be optimizing for the right target and require more nuanced ways to assess alignment. This is something I’m thinking about currently.
LLM-centric Approaches— All of these methods produce outputs that should eventually be fed into a central LLM scientist making decisions about experiments. How much of the analysis of characterization should be handled by bespoke models like the ones described above, how much can be handled by the LLM? Maybe just the basic ML classification of properties is enough for LLMs to reason about structural implications. Maybe autonomous characterization should be focused on enabling agents to use all the traditional tools scientists currently use, under the assumption that long-context memory and reasoning will be solved. The asymmetry of verification for generating reasonable structures that match up with spectra is excellent for many spectroscopic techniques. Those where calculating spectra from structure is expensive can be improved with better ML surrogates. Maybe this implies RL + SFT can enable LLMs to complete the entire task. My intuition is that generative models and neural networks that respect the nature of materials, like GNNs and equivariant diffusion, will still be necessary for building AI that can interpret characterization and feed useful information back into the loop. This is what I’ll be thinking about for a while.
Hardware for Autonomous Characterization — Another interesting problem in this direction is building new characterization instrumentation capable of high-throughput and autonomous operation. Autonomous operation might be a last-mile thing, similar to it being very hard to build a robot plumber—but hardware designed for high-throughput measurement with fairly consistent samples/workflows (the ones suitable for future autonomous labs) is feasible. Bringing synchrotron characterization data to autonomous materials discovery either by building online high-throughput hardware at synchrotron endstations (cool proof-of-concept) or lab-scale versions of these advanced techniques is also worth considering.
Closing Thoughts
I truly believe we’re on the cusp of some amazing things in AI for materials discovery and the impacts of materials abundance will bring about huge improvements to the world—so much so that I’m betting the start of my career on it while my friends at Stanford are chasing quant, finance, saas startups, or the current hot topic in LLMs.
I hope this essay achieves a few things:
- For those outside the direct AI for materials sphere, I hope there was valuable insight into the shape of the frontier of AI-driven materials discovery that I’ve picked up through lots of reading, research, and conversations with cool people over the past few months.
- For the scientists in the ML for materials community, I hope to spark some thoughts on how the rapid rise of LLMs has redefined the scope of AI in materials research. Not just inverse design, representation learning, or bayesian optimization-driven autonomous labs, but AI scientists that can intelligently reason through and carry out the entire scientific method.
- For those at the materials discovery neolabs, I hope to have brought some attention to the bottleneck of characterization and how unlocking characterization-driven structural insights in the loop is one of the strongest levers to getting real-world results.
My views will continue to change and this essay is by no means definitive, but I do believe writing is the best way to test your own understanding and putting your views out there is the fastest way to get proven wrong.
I’d love feedback from anyone with thoughts on any of the topics mentioned or ML for materials more broadly. I’m also open to research collaborators—I’ll be spending my summer working full-time on research projects in the space, so if you find ML for materials characterization interesting or would like to collaborate on other projects, feel free to reach out!
I’d like to thank Sam and Elena from ReGen, Andy Anker, Mike Mettler, Johannes Voss, Jehad Abed, Agniv Sarkar, James Liu, Joe Li, and Danica Sun for their valuable feedback on this essay.
Footnotes
[1] “Figure 2. (a) Contour plot of operando XRD patterns obtained on a NCM811/Li cell, together with structural refinement results (b, c) as well as differential lattice parameter (d) and capacity curves (e). The changes in unit cell volume and lattice parameters a and c versus the lithium content are shown in (b) and (c), respectively.
Figure 2b shows the decrease in unit cell volume from 101.38(1) to 94.26(2) Å3 (at x(Li) = 0.25) with delithiation. This contraction occurs in a nonlinear manner. At the beginning of the charge cycle, the unit cell volume decreases slowly until a lithium content of x(Li) ≈ 0.5 is reached. Then, the volume decreases much faster, and for x(Li) < 0.4, it decreases virtually linearly and steeply with lithium content until the end of charge. The unit cell volume is determined by the lattice parameters a and c, which are affected by the intralayer spacing and slab heights, respectively. As can be seen from Figure 2c, the a lattice parameter exhibits an almost linear drop from 2.8661(1) to 2.8211(1) Å (at x(Li) = 0.5) and then levels off at 2.8153(1) Å (at x(Li) = 0.25). Interestingly, the initial decrease in unit cell volume bears some resemblance to the initial decrease in a. Although the absolute changes are significantly smaller than those observed for c, the a lattice parameter contributes to the cell volume by the second power (V = a2c sin π/3), and therefore determines the magnitude of the changes in this region (at high lithium content). The c lattice parameter exhibits much more complex behavior, showing an initial increase from 14.249(1) to 14.469(1) Å (at x(Li) = 0.6), followed by a broad maximum and finally a steep decrease to 13.732(2) Å (at x(Li) = 0.25). The latter drop is strongly reminiscent of that observed for the unit cell volume at x(Li) ≤ 0.5. This feature, together with the fact that the lattice parameter a barely changes in this region, establishes that the unit cell volume is mainly controlled by the c lattice parameter when x(Li) ≤ 0.5.”
[2] The optimization approach may work for some problems. Materials problems can be put on a spectrum from being a search problem to being a research problem. Some materials, like polymers and MOFs, are more suited for this high throughput trial and error approach. And indeed, data-driven methods can sometimes propose suggestions that escape human chemical intuition. But for a broader set of highly relevant materials relevant to a material abundant future—batteries, catalysts, solar cells, semis, fusion walls—progress requires not only new suggestions, but extensive characterization and the insight derived from it.
As for the limitations of characterization thus far in autonomous labs, they’re limited to a few more easily/quantitatively interpretable techniques like XRD which are limited to crystal structures (when many important systems are not perfectly crystalline), and significant work is required to train models to interpret other techniques. Additionally, the measurements are still used mostly as a signal for a synthesis parameter optimization algorithm, not to understand mechanisms in complex, functional materials.
[3] AI for bio has progressed much faster than materials, perhaps in part because it received more early attention from investors and the broader tech world due to its more palpable benefits, but also due to technical reasons. Although many of the intuitions and even technical frameworks carry over, bio has much larger and more trustworthy datasets, more standardized/automatable experimental workflows, and a data layer that can more easily capture complete information about a sample (e.g. ATCG). Bio also has illumina sequencers generating a super low cost-per-bit, as well as “compile uniformity” just like codegen—something that just doesn’t apply for materials. The mapping from representation (data) to function is much cleaner in bio than materials. In materials, performance is usually not a function of composition alone, or even crystal structure alone, but of synthesis history, defects, interfaces, microstructure, metastability, and operating environment. In other words, the object you want to predict is often not a single material but a processing-dependent physical system. All these differences make the ceiling of data-driven methods lower for materials than bio, hence the emphasis here on the scientific method and unlocking mechanistic insight.